If you find mistakes or suggestions, please report them at issues.
We wanted to train new models for R10.4.1 for use with f5c. f5c has no implementation for training new models. Given f5c relies on nanopolish models, we use nanopolish for training such models. The process of figuring out the nanopolish training for R10.4.1 was quite challenging and required considerable effort. With great help from Jared Simpon, we were able to figure it out. Hence, we have briefly documented the process we used to train R10.4.1 with nanopolish, and hope it can benefit someone else.
We sequenced the human methylated and non-methylated (WGA) DNA datasets from zymo dna methylation standards (D5013). One PromethION flowcell was used for each dataset and we obtained ~7X depth for the non-methylated and ~9X for the methylated. The whole genome amplified non-methylated data with no base modifications is used for training the nucleotide model. Both methylated (only CG methylated in this Zymo dataset) and non-methylated are needed for training a methylation model in CpG context.
In this example, We will be using BLOW5 files to train as BLOW5 is much faster and more convenient. You can get the nanopolish r10 branch with BLOW5 support from here (commit used ce29c2da, remember to checkout to r10 branch) and compile it. The binary compiled on Ubuntu 16 is available under slow5-nanopolish-train/ubuntu16 here and will work if your Linux distribution is recent. For older Linux distributions, try the binary under slow5-nanopolish-train/centos7.
The data is available on ENA under PRJEB64592.
Nucleotide model
- The base-model provided by ONT for dna_r10.4.1_e8.2_400bps was used as the base model. The current level values in the ONT model are normalised. We convert to nanopolish model format as below (file
r10.4.1_400bps.nucleotide.9mer.modelavailable here):#ont_model_name r10_180mv_450bps_9mer #kit r10_450bps #strand template #k 9 #alphabet nucleotide kmer level_mean level_stdv sd_mean sd_stdv weight AAAAAAAAA 54.2049064106214 3.0 1 1 1 1 AAAAAAAAC 58.590797271313 3.0 1 1 1 1 AAAAAAAAG 55.9520679998075 3.0 1 1 1 1 AAAAAAAAT 58.4335200897368 3.0 1 1 1 1 AAAAAAACA 63.6599592820753 3.0 1 1 1 1Though we are training a model for dna_r10.4.1_e8.2_400bps, note that in the model above we have r10_180mv_450bps_9mer as the model, to trick nanopolish train to work. For converting the levels from ONT model to pA above, we used
level*23.027081308793+96.631070539403, with mean and std constants calculated using sigtk pa and datamash on a R10.4.1 dataset. Note that level_stdv is set to 3.0 for all kmers to start with. Other columns such as sd_mean are unused, so leave them as 1. - Assume we have the non-methylated dataset in a BLOW file called
nonmeth_reads.blow5. Basecall the BLOW5 file with super accuracy through buttery-eel using Guppy:buttery-eel -g /install/ont-guppy-6.4.2/bin/ --config dna_r10.4.1_e8.2_400bps_sup.cfg --device 'cuda:all' -i nonmeth_reads.blow5 -q 10 -o nonmeth.fastq --port 5555This will create two files named
nonmeth.pass.fastqandnonmeth.fail.fastq. We will usenonmeth.pass.fastqcontaining passed reads (reads with mean qscore > 10). - Now let us use Minimap2 to map. We Used hg38 with no alternate contigs as the reference:
minimap2 -ax map-ont /genome/hg38noAlt.fa --secondary=no -t20 nonmeth.pass.fastq | samtools sort - > nonmeth_pass.bam samtools index nonmeth_pass.bam - Before running nanopolish train, we need to replace the few ‘M’ IUPAC bases in the hg38noAlt.fa with ‘N’ bases. Otherwise, Nanopolish will error out as ‘M’ is the symbol used to represent methylated bases in nanopolish. Assume that the patched reference is named
hg38noAlt_M_replaced_N.faand the compressed version namedhg38noAlt_M_replaced_N.fa.gzcan be download from here. Now let us do the training.slow5-nanopolish-train/nanopolish index nonmeth.pass.fastq --slow5 nonmeth_reads.blow5 slow5-nanopolish-train/nanopolish train -r nonmeth.pass.fastq -g hg38noAlt_M_replaced_N.fa -b nonmeth_pass.bam -t 72 --train-kmers=all --input-model-filename ./r10.4.1_400bps.nucleotide.9mer.model -d trained_nuc_models/If training is successful, you should see files like
r10_450bps.nucleotide.9mer.template.roundX.modelundertrained_nuc_models(they are uploaded here). They should look like:#model_name r10_450bps.nucleotide.9mer.template.model #kit r10_450bps #strand template #alphabet nucleotide AAAAAAAAA 54.2003 1.99094 1 1 AAAAAAAAC 57.2213 2.59858 1 1 AAAAAAAAG 55.7136 2.52011 1 1 AAAAAAAAT 57.2297 2.71002 1 1Also, there will files like
r10_450bps.nucleotide.9mer.template.roundX.summmary.tsv. Make sure in those files, theis_trainedcolumn is set to 1:model_short_name kmer num_matches num_skips num_stays num_events_for_training was_trained trained_level_mean trained_level_stdv r10_450bps.t CAAAAAAAA 0 0 0 1000 1 56.42 1.82 r10_450bps.t TTCAAAAAA 0 0 0 1000 1 60.69 2.46 r10_450bps.t AATAAAAAA 0 0 0 1000 1 57.26 3.36 r10_450bps.t CCTAAAAAA 0 0 0 1000 1 60.58 2.35 r10_450bps.t AAAAAAAAA 0 0 0 1000 1 54.20 1.99If all of them are 0, that means they were not successfully trained. By default, nanopolish will require at least 100
num_events_for_training, so unless you have good coverage for all k-mers, some of the k-mers will not be trained. In our case, we had around ~7X depth for the human genome and 99.42% of the k-mers were trained (260631 of 262144), but not all. If the outputs are empty or if all k-mers are not trained (which happened to us at the beginning), it could be due to multiple reasons. I do not remember much of those now, but these issues may help with debugging: #825, #761, #1059, #1064. - Then we used the last model trained in step 4 above (
trained_nuc_models/r10_450bps.nucleotide.9mer.template.round4.model) as the input to train 10 more rounds.slow5-nanopolish-train/nanopolish train -r nometh.pass.fastq -g hg38noAlt_M_replaced_N.fa -b nometh_pass.bam -t 72 --train-kmers=all --input-model-filename trained_nuc_models/r10_450bps.nucleotide.9mer.template.round4.model -d trained_nuc_models_more/ --rounds 10The created model called
trained_nuc_models_more/r10_450bps.nucleotide.9mer.template.round9.model(they are uploaded here) which looked like below, is the final model that we put as the inbuilt model for f5c-v1.2-beta:#model_name r10_450bps.nucleotide.9mer.template.model #kit r10_450bps #strand template #alphabet nucleotide AAAAAAAAA 53.9989 3.83325 1 1 AAAAAAAAC 55.769 3.20809 1 1 AAAAAAAAG 56.4699 5.17555 1 1 AAAAAAAAT 56.385 3.47242 1 1 AAAAAAACA 59.1981 4.42718 1 1 AAAAAAACC 62.2545 5.09254 1 1 AAAAAAACG 60.252 4.37649 1 1 AAAAAAACT 62.0946 4.71852 1 1 AAAAAAAGA 89.6554 29.6328 1 1 AAAAAAAGC 63.4326 4.10237 1 1 AAAAAAAGG 59.7029 4.50327 1 1 AAAAAAAGT 62.8706 3.91294 1 1 - To determine how training went, we used f5c eventalign to align a set of hg2 reads (BLOW5 file available here) using the model file created after each training round. An example command that uses the last training round model:
f5c eventalign -x hpc-low -b hg2_subsample_guppy_6.4.2_hac_pass.bam -r hg2_subsample_guppy_6.4.2_hac_pass.fastq -g /genome/hg38noAlt.fa --slow5 hg2_subsample.blow5 --kmer-model trained_nuc_models_more/r10_450bps.nucleotide.9mer.template.round9.model > event.tsvAt the end, f5c prints some alignment stats to the standard error:
total entries: 482419, qc fail: 900, could not calibrate: 318, no alignment: 3924, bad reads: 0The plots below show how each training round improved the number of successful alignments and reduced the number of failed alignments (failed alignments = qc fail + could not calibrate + no alignment; successful alignments = total entries - failed alignments).


Methylation model
- We need both non-methylated data (used for nucleotide model training above) and methylated data for training a model for methylation calling. We already basecalled the non-methylated data at step 1 above. Now let us basecall the methylated dataset.
buttery-eel -g /install/ont-guppy-6.4.2/bin/ --config dna_r10.4.1_e8.2_400bps_sup.cfg --device 'cuda:all' -i meth_reads.blow5 -q 10 -o meth.fastq --port 5555 - Now combine passed reads from the non-methylated dataset and the methylated dataset into one single fastq file.
cat nonmeth.pass.fastq meth.pass.fastq > positive_and_negative_pass.fastq - Merge the methylated and non methylated BLOW5 files into one single file:
slow5tools merge meth_reads.blow5 nonmeth_reads.blow5 -o merged.blow5 - Now we need to create a methylated reference - one with all CGs changed into MG. For this, we use the
hg38noAlt_M_replaced_N.fawe created in step 4 above.# make a copy with chr renamed to chr_meth sed 's/chr/chr_meth_/g' hg38noAlt_M_replaced_N.fa > hg38noAlt_chr_renamed_to_meth.fa # use seqtk to create single lined FASTA and replace all CG sites with MG. seqtk seq -l0 hg38noAlt_chr_renamed_to_meth.fa | sed 's/CG/MG/g' > hg38noAlt_chr_renamed_to_meth_methylated_cpg.faM is the nanopolish symbol for methylated bases. The boundaries (C’\n’G) will not be properly replaced if you use a multiline FASTA, that is why seqtk is used above to make it a single-lined FASTA.
- Map the methylated reads to the renamed reference
hg38noAlt_chr_renamed_to_meth.fawe created in the previous step above (not thehg38noAlt_chr_renamed_to_meth_methylated_cpg.fa)minimap2 -ax map-ont hg38noAlt_chr_renamed_to_meth.fa--secondary=no -t20 meth.pass.fastq | samtools sort - > meth_pass.bam samtools index meth_pass.bam - Merge the BAM file from non-methylated data (
nonmeth_pass.bamwe created before for in step 3 above) and the BAM file from methylated data (meth_pass.bam) into a single BAM file.samtools merge merged.bam nonmeth_pass.bam meth_pass.bam samtools index merged.bam - Now create the hybrid reference to be later used with nanopolish.
seqtk seq -l0 hg38noAlt_M_replaced_N.fa > hg38noAlt_tmp.fa cat hg38noAlt_tmp.fa hg38noAlt_chr_renamed_to_meth_methylated_cpg.fa > hg38noAlt_hybrid.fa - Now we need to create a base methylated model for nanopolish. What we did was, we took the
r10_450bps.nucleotide.9mer.template.round9.modelnucleotide model which was trained above, created a new model with ‘M’ in the alphabet and put the same level_mean and level_stdv as for the corresponding ‘C’ counterparts. This file is named asr10.4.1_400bps.cpg.9mer.modelwhich is available here.#model_name r10_450bps.cpg.9mer.template.model #kit r10_450bps #strand template #k 9 #alphabet cpg kmer level_mean level_stdv sd_mean sd_stdv weight AAAAAAAAA 53.998900 3.833250 1 1 1 1 AAAAAAAAC 55.769000 3.208090 1 1 1 1 AAAAAAAAG 56.469900 5.175550 1 1 1 1 AAAAAAAAM 55.769000 3.208090 1 1 1 1 AAAAAAAAT 56.385000 3.472420 1 1 1 1 AAAAAAACA 59.198100 4.427180 1 1 1 1 AAAAAAACC 62.254500 5.092540 1 1 1 1 AAAAAAACG 60.252000 4.376490 1 1 1 1 AAAAAAACM 62.254500 5.092540 1 1 1 1 AAAAAAACT 62.094600 4.718520 1 1 1 1 AAAAAAAGA 89.655400 29.632800 1 1 1 1 AAAAAAAGC 63.432600 4.102370 1 1 1 1 AAAAAAAGG 59.702900 4.503270 1 1 1 1 AAAAAAAGM 63.432600 4.102370 1 1 1 1 AAAAAAAGT 62.870600 3.912940 1 1 1 1 AAAAAAAMA 59.198100 4.427180 1 1 1 1 AAAAAAAMC 62.254500 5.092540 1 1 1 1 AAAAAAAMG 60.252000 4.376490 1 1 1 1 AAAAAAAMM 62.254500 5.092540 1 1 1 1 AAAAAAAMT 62.094600 4.718520 1 1 1 1 AAAAAAATA 58.516100 4.245120 1 1 1 1 AAAAAAATC 60.291300 4.159900 1 1 1 1 AAAAAAATG 60.182800 4.553570 1 1 1 1 AAAAAAATM 60.291300 4.159900 1 1 1 1 AAAAAAATT 59.724500 3.398340 1 1 1 1 AAAAAACAA 101.404000 18.021500 1 1 1 1 AAAAAACAC 108.078000 12.647600 1 1 1 1 AAAAAACAG 100.971000 18.010800 1 1 1 1 AAAAAACAM 108.078000 12.647600 1 1 1 1 AAAAAACAT 107.755000 11.699300 1 1 1 1 AAAAAACCA 101.966000 20.410900 1 1 1 1 AAAAAACCC 109.189000 13.997700 1 1 1 1 - Now let us use nanopolish to train a methylation model.
slow5-nanopolish-train/nanopolish index positive_and_negative_pass.fastq --slow5 merged.blow5 slow5-nanopolish-train/nanopolish train -r positive_and_negative_pass.fastq -g hg38noAlt_hybrid.fa -b merged.bam -t 72 --train-kmers=all --input-model-filename r10.4.1_400bps.cpg.9mer.model -d trained_meth_models --rounds 10 - The created
trained_meth_models/r10_450bps.cpg.9mer.template.round9.model(available for download here) looked like below :#model_name r10_450bps.cpg.9mer.template.model #kit r10_450bps #strand template #alphabet cpg AAAAAAAAA 53.7537 3.65572 1 1 AAAAAAAAC 56.2305 4.32346 1 1 AAAAAAAAG 57.3002 5.67074 1 1 AAAAAAAAM 62.9934 8.11058 1 1 AAAAAAAAT 56.7203 4.17047 1 1 AAAAAAACA 58.8781 4.15414 1 1 AAAAAAACC 62.5381 5.1778 1 1 AAAAAAACG 60.3428 5.31056 1 1 AAAAAAACM 63.1525 4.73173 1 1 AAAAAAACT 61.8409 4.19311 1 1 AAAAAAAGA 92.1803 31.218 1 1 AAAAAAAGC 63.5452 4.65351 1 1 AAAAAAAGG 61.4242 5.45205 1 1 AAAAAAAGM 68.0082 6.17807 1 1 AAAAAAAGT 62.7944 4.12666 1 1 AAAAAAAMA 59.1981 4.42718 1 1 AAAAAAAMC 62.2545 5.09254 1 1 AAAAAAAMG 61.1943 3.33436 1 1 AAAAAAAMM 62.2545 5.09254 1 1 AAAAAAAMT 62.0946 4.71852 1 1 AAAAAAATA 58.8587 4.67559 1 1 AAAAAAATC 60.3588 4.00821 1 1 AAAAAAATG 60.477 5.42577 1 1 AAAAAAATM 61.3608 3.74302 1 1 AAAAAAATT 59.6004 3.20899 1 1 AAAAAACAA 99.7551 17.5451 1 1 AAAAAACAC 107.298 12.0526 1 1 AAAAAACAG 101.281 17.5249 1 1 AAAAAACAM 109.249 13.1459 1 1 AAAAAACAT 107.25 11.8888 1 1Make sure that
r10_450bps.cpg.9mer.template.round9.summmary.tsvhas the column “was_trained” set to 1 for k-mers with CG and MG. If the outputs are empty or if all k-mers are not trained (which happened to us at the beginning), it could be due to multiple reasons. I had to manually replace the inbuilt .inl model in nanopolish with values from the trained nucletide modeltrained_nuc_models_more/r10_450bps.nucleotide.9mer.template.round9.model(see here). If you are using the latest r10 branch from https://github.com/hiruna72/nanopolish/ or the binary download from here this is already done for you. These issues may help debugging if you are having troubles: #825, #761, #1059, #1064. - To see how the training went, I used f5c call-methylation with trained methylation models from each round. For this, I used a ~30X chr22 HG2 dataset.
f5c call-methylation -x hpc-low -b hg2_prom_lsk114/chr22/hac/reads.bam -r hg2_prom_lsk114/chr22/hac/reads.fastq -g /genome/hg38noAlt.fa --slow5 hg2_prom_lsk114/chr22/PGXX22394_reads_chr22.blow5 --kmer-model trained_nuc_models_more/r10_450bps.nucleotide.9mer.template.round9.model --meth-model trained_meth_models/r10_450bps.cpg.9mer.template.round9.model > meth.tsv f5c meth-freq -i meth.tsv -s > meth-freq.tsvThen I used the methylation comparison method described here. For your convenience, I have put the necessary scripts and the data to be downloaded under
eval/meth/from here.echo -e "chromosome\tstart\tend\tnum_motifs_in_group\tcalled_sites\tcalled_sites_methylated\tmethylated_frequency\tgroup" > meth-freqchr22.tsv grep chr22 meth-freq.tsv >> meth-freqchr22.tsv python3 eval/meth/compare_methylation.py eval/meth/chr22.tsv meth-freqchr22.tsv > bul_vs_f5c.tsv R --save < eval/meth/plot_methylation.RThe data
eval/meth/chr22.tsvis bisulphite data for chr22 of HG2 curated from here. How the called number of sites (N) and correlation (r) change with each training round are as below: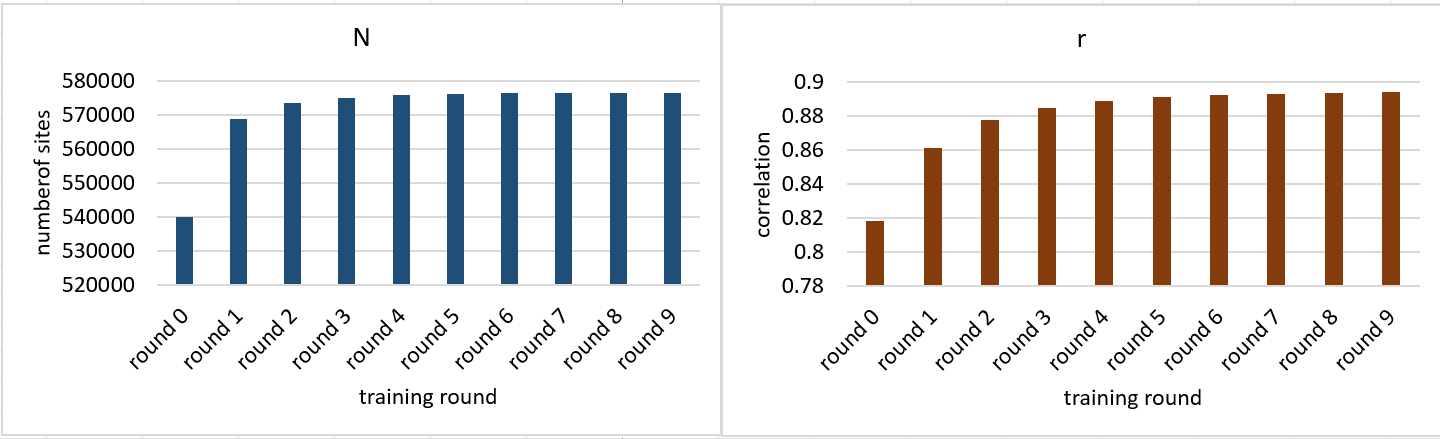
The correlation plot from
trained_meth_models/r10_450bps.cpg.9mer.template.round9.modelis as follows: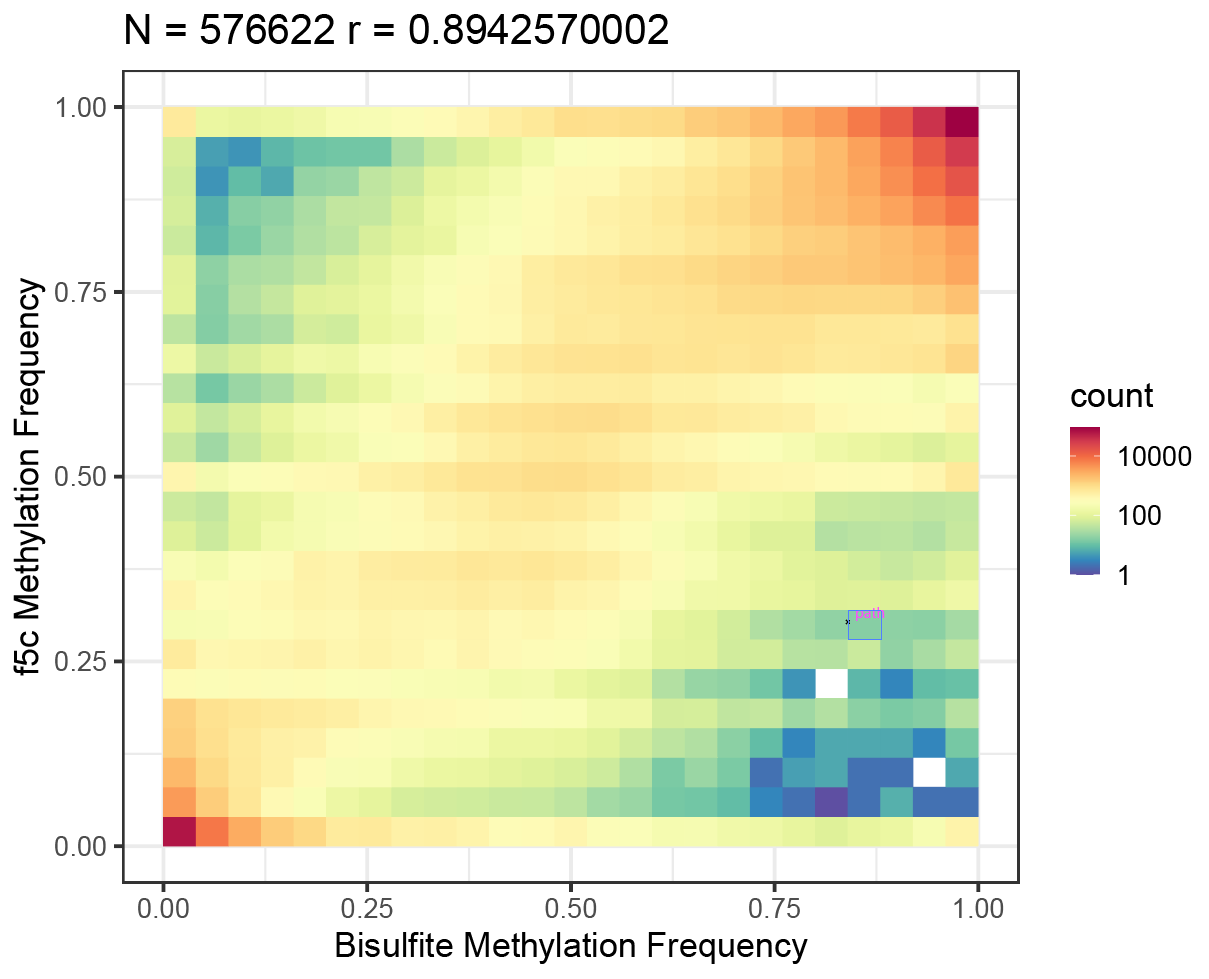
The
trained_meth_models/r10_450bps.cpg.9mer.template.round9.modelwas used as the inbuilt model for f5c-v1.2-beta.
Caveats and Possible Improvements
-
As we used minimal data to train (the coverage for human methylated and non-methylated datasets was <10X coverage), the models are not perfect. Some k-mers did not even have enough coverage to train (<100 events)). But the good thing is, now anyone who has better data can use the models we generated as the base models (
trained_nuc_models_more/r10_450bps.nucleotide.9mer.template.round9.modelas the base nucleotide model andtrained_meth_models/r10_450bps.cpg.9mer.template.round9.modelas the base methylation model) and train futher. -
The sequencing we did was for 400bps speed and thus the models may not work well for 260bps. Anyway, ONT is deprecating 260bps I heard.
-
Nanopolish train also has this
--bedmethyl=FILEoption that can infer the presence/absence of methylation sites using FILE as a map, which could be a useful feature that I have yet to explore. -
Many meta parameters such as those for event detection, scaling and HMM structure used for R10 are the same as for R9. These parameters could be optimised for R10 but haven’t got time to explore them. Thus unless a lot of more work is done, it is not possible to say whether what f5c currently gives for R10 is the best that can be achieved from nanopolish-style models.